James Stroud wrote:
Hello,
I have a 2.45 A structure with an average b factor of 50.6. A region I
am particularly interested in has an average b factor of 87. At what
point do I say that the region is "disordered"? Does it come down to
maps? If I have reasonable simulated omit maps but the b factor is 87,
how much confidence can I have about my interpretation of the maps?
James
I think the traditional definition of "disordered" is that you don't see
it in a map contoured at 1 sigma. What kind of map seems to depend on
your particular scientific pedigree and the epoch of crystallographic
convention in which you live, such as the 2fo-fc era. The 3fo-2fc clan,
or the current maximum-likelihood era of 2mFo-DFc. However, if the
atoms are particularly important to you, you might not call them
"disordered" until they don't show up in a Fo-Fc map contoured at 0.5 sigma.
There does not appear to be a clear definition. In fact, what
crystallographers call "disordered" might be called "high resolution" in
some other technique. After all, you know the atoms are in the unit
cell somewhere, so your resolution is already ~10x better than the best
visible light microscopes. Hence, the B-factor is somehow connected to
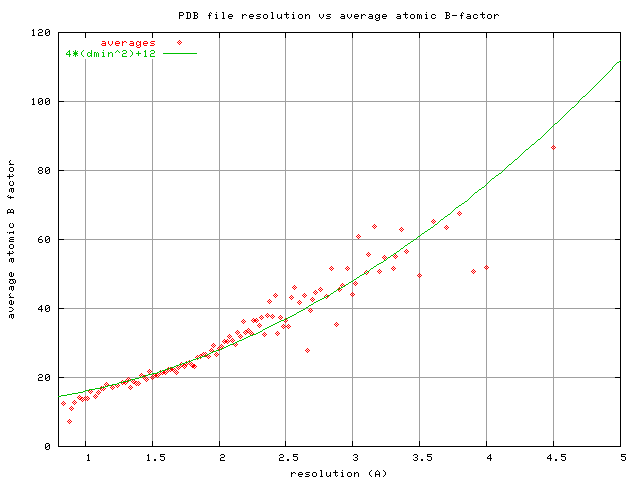
resolution. I once did a survey of the PDB (Equation 4 in Holton JSR
2009) plotting the average atomic B-factor of all PDB files with a given
stated "RESOLUTION" listed in the file. The plot (which I did not show
in the paper) is here:
http://bl831.als.lbl.gov/~jamesh/pickup/reso_vs_avgB.png
One possible origin of the average general trend (B = 4*d^2+12 where d
is the resolution) is because a B-factor Gaussian has a full-width at
half-max roughly given by B = 14.2*fwhm^2 and if we consider that B ~
4*d^2 and sqrt(14.2/4) ~ 2, this implies that 2*fwhm ~ d, or that the
"feature size" at a d-spacing of d is ~d/2 (picture a sine wave with
full-cycle period d).
The minimum average B value of 12 for low d could arise because a carbon
atom at rest has the width of a B-factor Gaussian with B ~ 12 (0.9 A),
and since the actual B-factor Gaussian is convoluted with this shape, B
values smaller than 12 have vanishingly small influence on the electron
density. ... Maybe.
I'm sure there are other explanations for the origins of this trend, and
I admit there are plenty of PDBs that do not obey it. (It is an average
trend) But, this expression does suggest how one might relate the
B-factor of an atom to an "effective resolution". That is, atoms with
high B-factors do not contribute significantly to high-angle spots, but
atoms with low B-factors do. So, one could say that your overall
average B=50.6 indicates an effective resolution of 3.1 A and your "bad"
region has an effective resolution of 4.3 A. Now, your observed
resolution is 2.45 A, which implies that your data are better than
average, or perhaps your average B is being pulled up by the high-B
region?
Nevertheless, I still find it useful to convert B-factors into
equivalent d-spacings because it is easier to think about how good
electron density looks in terms of resolution. That is, your electron
density is a sum of a 4.3 A structure (your "bad" region) and a 2.45 A
structure (your atoms with B ~ 36), which leads to an average overall B
of 50.6. Therefore, your "disordered" region has electron density
similar to that of a typical 4.3 A structure, and the kinds of
conclusions you can make from that part of the structure will be similar
to what you could conclude if the overall structure were 4.3 A. Sorry.
This is probably not what you wanted to hear.
Interestingly, however, protein crystals that respond well to
dehydration treatments (such as with the Proteros/Rigaku FMS) tend to be
cases where one molecule in the ASU has significantly higher B than
another. After dehydration, the "loose" molecule orders up (sometimes).
-James Holton
MAD Scientist
{kind=link}