On 11/20/12 11:30 AM, Ali Alizadeh wrote:
Dear All users
I want my energy minimization of my system is converged to considered
value, when i do it,
It is converged because it reachs to Fmax and i see this message :
Steepest Descents converged to Fmax < 10 in 31702 steps
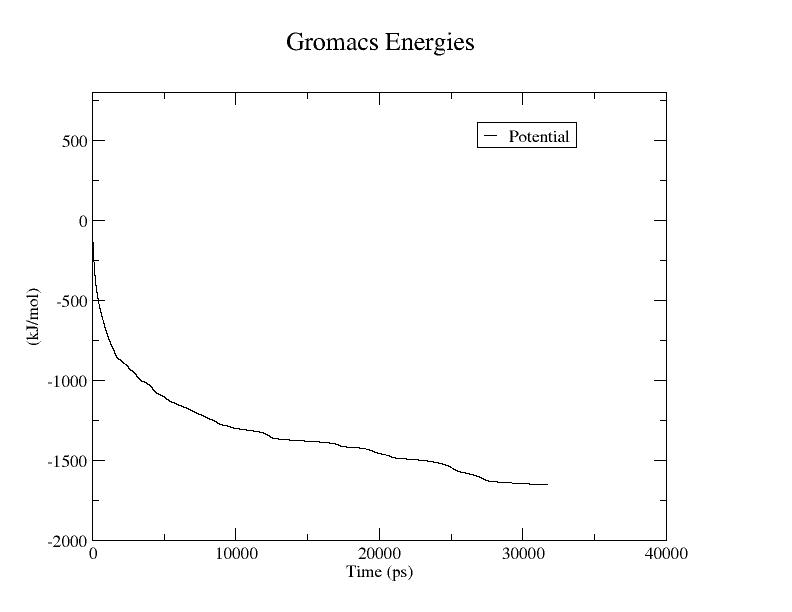
Potential Energy = -1.6531438e+03
Maximum force = 9.8014317e+00 on atom 162
Norm of force = 2.9060462e+00
my result:
http://alichemical.persiangig.com/document/em.jpg
When i represent profile of potential energy by g_energy, i can't see
my considered results,
I should reach this result that this link is related to it,
http://alichemical.persiangig.com/document/11.jpg
Justin wrote:
The first plot shows the result of EM, the second is the
result of a relatively
long MD simulation. I would not expect them to show the same
result. Further,
it appears that the second plot pertains to the
configurational energy per water
molecule (based on magnitude), while the first simply measures
the potential
energy of the whole system. Since the system is homogeneous,
you can use the
-nmol option of g_energy to divide the energy terms by the
number of molecules.
-Justin
Dear Justin
Thank you for your reply,
1- In the first plot, unit of y axis is kj/mol, and you said above i
should use -nmol, i confused, Why is the unit of y axis kj/mol
without using -nmol?
Because that's the energy unit Gromacs always uses. The energy given is kJ per
mole of equivalent systems. In the second plot shown above, the magnitude of
the energy suggests kJ per mole of individual molecules.
2- I want do EM so that energy of my system reaches to considered
value, but i never reach it exactly, what's wrong?
You set an EM tolerance of 10 kJ/(mol-nm), which is a force, and mdrun achieved
that. You can't ask an EM algorithm to produce the same result as a long-time
MD run, as is shown above.
-Justin
--
========================================
Justin A. Lemkul, Ph.D.
Research Scientist
Department of Biochemistry
Virginia Tech
Blacksburg, VA
jalemkul[at]vt.edu | (540) 231-9080
http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin
========================================
--
gmx-users mailing list gmx-users@gromacs.org
http://lists.gromacs.org/mailman/listinfo/gmx-users
* Please search the archive at
http://www.gromacs.org/Support/Mailing_Lists/Search before posting!
* Please don't post (un)subscribe requests to the list. Use the
www interface or send it to gmx-users-requ...@gromacs.org.
* Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
{kind=link}
{kind=link}