Dear Bob, dear Angel, In relation with the P2N file format (previous messages), I also have a question regarding the PDB file format & Jmol.
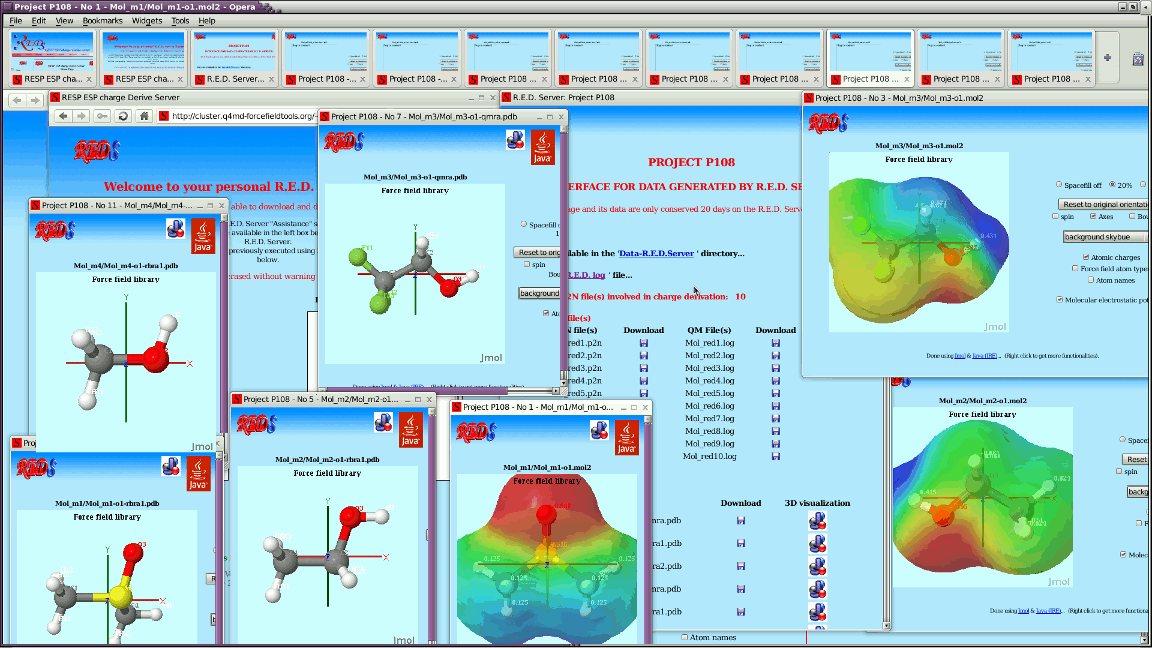
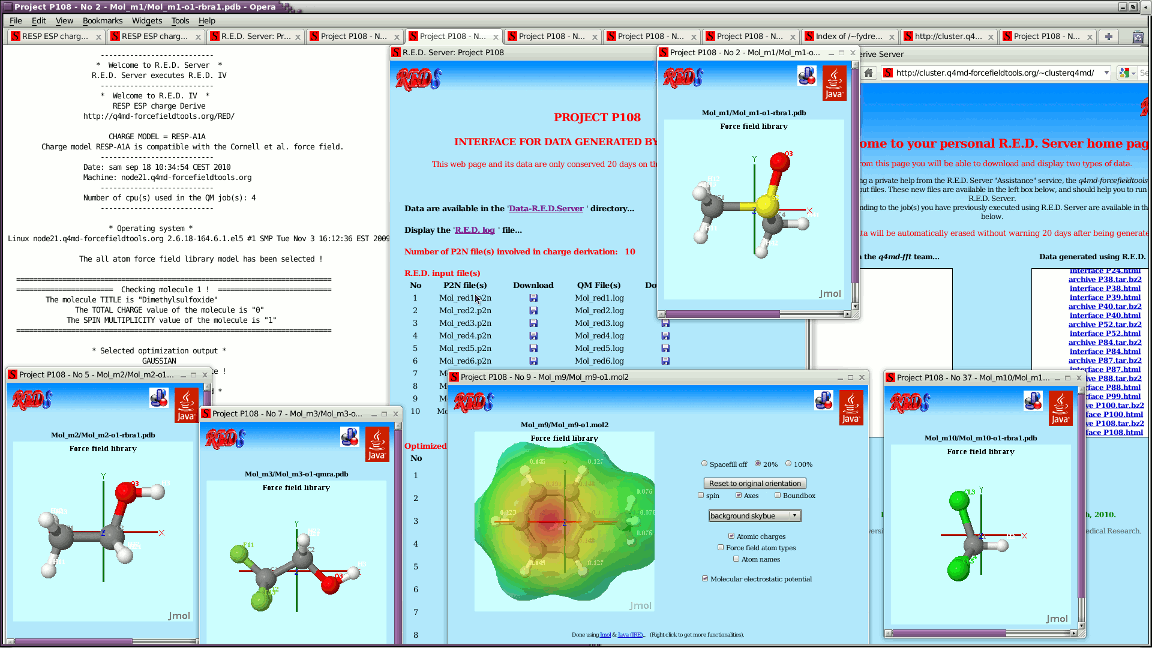
I wrote two Jmol applets @ http://cluster.q4md-forcefieldtools.org/~ucpublic1/javaappletpdb-1.html which loads http://cluster.q4md-forcefieldtools.org/~ucpublic1/Mol_m1-o1-qmra.pdb i.e. ATOM 1 CA1 BNZ 1 0.5624 1.2144 0.3611 C ATOM 2 H1 BNZ 1 0.9988 2.1568 0.6413 H ATOM 3 CB2 BNZ 1 -0.7794 1.1461 0.0200 C ATOM 4 H2 BNZ 1 -1.3843 2.0354 0.0356 H ATOM 5 CC3 BNZ 1 -1.3418 -0.0683 -0.3411 C ATOM 6 H3 BNZ 1 -2.3830 -0.1213 -0.6057 H ATOM 7 CD4 BNZ 1 -0.5624 -1.2144 -0.3611 C ATOM 8 H4 BNZ 1 -0.9988 -2.1568 -0.6413 H ATOM 9 CE5 BNZ 1 0.7794 -1.1461 -0.0200 C ATOM 10 H5 BNZ 1 1.3843 -2.0354 -0.0356 H ATOM 11 CF6 BNZ 1 1.3418 0.0683 0.3410 C ATOM 12 H6 BNZ 1 2.3830 0.1213 0.6057 H and http://cluster.q4md-forcefieldtools.org/~ucpublic1/javaappletpdb-2.html which loads http://cluster.q4md-forcefieldtools.org/~ucpublic1/Mol_m2-o1-qmra.pdb i.e. ATOM 1 CA1 BNZ 1 0.5624 1.2144 0.3611 ATOM 2 H1 BNZ 1 0.9988 2.1568 0.6413 ATOM 3 CB2 BNZ 1 -0.7794 1.1461 0.0200 ATOM 4 H2 BNZ 1 -1.3843 2.0354 0.0356 ATOM 5 CC3 BNZ 1 -1.3418 -0.0683 -0.3411 ATOM 6 H3 BNZ 1 -2.3830 -0.1213 -0.6057 ATOM 7 CD4 BNZ 1 -0.5624 -1.2144 -0.3611 ATOM 8 H4 BNZ 1 -0.9988 -2.1568 -0.6413 ATOM 9 CE5 BNZ 1 0.7794 -1.1461 -0.0200 ATOM 10 H5 BNZ 1 1.3843 -2.0354 -0.0356 ATOM 11 CF6 BNZ 1 1.3418 0.0683 0.3410 ATOM 12 H6 BNZ 1 2.3830 0.1213 0.6057 As you can see http://cluster.q4md-forcefieldtools.org/~ucpublic1/javaappletpdb-1.html is ok, while http://cluster.q4md-forcefieldtools.org/~ucpublic1/javaappletpdb-2.html leads to ugly connections; I guess this because of the absence of chemical elements in Mol_m2-o1-qmra.pdb. Why does Jmol need chemical elements to recognize atom names? - Many small molecules are written in the PDB file format without these chemical elements - VMD and LEaP (for instance) do not need chemical elements to recognize atom names Do you think it would be possible that Jmol recognizes atom names without chemical elements? --- For each job, R.E.D. Server generates by now a Jmol-based graphical interface to display outputs generated by R.E.D. IV. See for instance: http://q4md-forcefieldtools.org/Tutorial/DNA-FFTopDB/P1.html & http://q4md-forcefieldtools.org/REDS/images/Screenshot1.gif http://q4md-forcefieldtools.org/REDS/images/Screenshot2.gif http://q4md-forcefieldtools.org/REDS/images/Screenshot3.gif The problem is that some of the applets generated for the PDB files present broken/bad atom connectivities because of the absence of chemical elements; we can obviously add chemical elements in our PDB files (no problem) - however doing so in the P2N file format might not be that straightforward... I continue now with these simple atom names ATOM 1 C1 BNZ 1 0.5624 1.2144 0.3611 ATOM 2 H1 BNZ 1 0.9988 2.1568 0.6413 ATOM 3 C2 BNZ 1 -0.7794 1.1461 0.0200 ATOM 4 H2 BNZ 1 -1.3843 2.0354 0.0356 ATOM 5 C3 BNZ 1 -1.3418 -0.0683 -0.3411 ATOM 6 H3 BNZ 1 -2.3830 -0.1213 -0.6057 ATOM 7 C4 BNZ 1 -0.5624 -1.2144 -0.3611 ATOM 8 H4 BNZ 1 -0.9988 -2.1568 -0.6413 ATOM 9 C5 BNZ 1 0.7794 -1.1461 -0.0200 ATOM 10 H5 BNZ 1 1.3843 -2.0354 -0.0356 ATOM 11 C6 BNZ 1 1.3418 0.0683 0.3410 ATOM 12 H6 BNZ 1 2.3830 0.1213 0.6057 no problem: atom connectivites are correct in a Jmol applet even in the absence of chemical elements; it also works for http://q4md-forcefieldtools.org/Tutorial/DNA-FFTopDB/P1/javaappletpdb-6.html where atom names are not that simple. Consequently, this means that Jmol can already recognize some atom names without chemical elements. Is it not possible to extend this feature to more general atom names? thanks a lot regards, Francois ------------------------------------------------------------------------------ Centralized Desktop Delivery: Dell and VMware Reference Architecture Simplifying enterprise desktop deployment and management using Dell EqualLogic storage and VMware View: A highly scalable, end-to-end client virtualization framework. Read more! http://p.sf.net/sfu/dell-eql-dev2dev _______________________________________________ Jmol-users mailing list Jmol-users@lists.sourceforge.net https://lists.sourceforge.net/lists/listinfo/jmol-users
{kind=link}
{kind=link}
{kind=link}